When a generic drug hits the market, you might assume it’s just a cheaper copy of the brand-name version. But behind that simple label is a rigorous scientific process designed to prove it works the same way in your body. That process is called bioequivalence testing. And it comes in two main forms: in vivo and in vitro. One happens inside the human body. The other happens in a lab dish. Knowing when each is used-and why-helps explain why some generics are approved quickly while others take years.
What Is Bioequivalence Testing and Why Does It Matter?
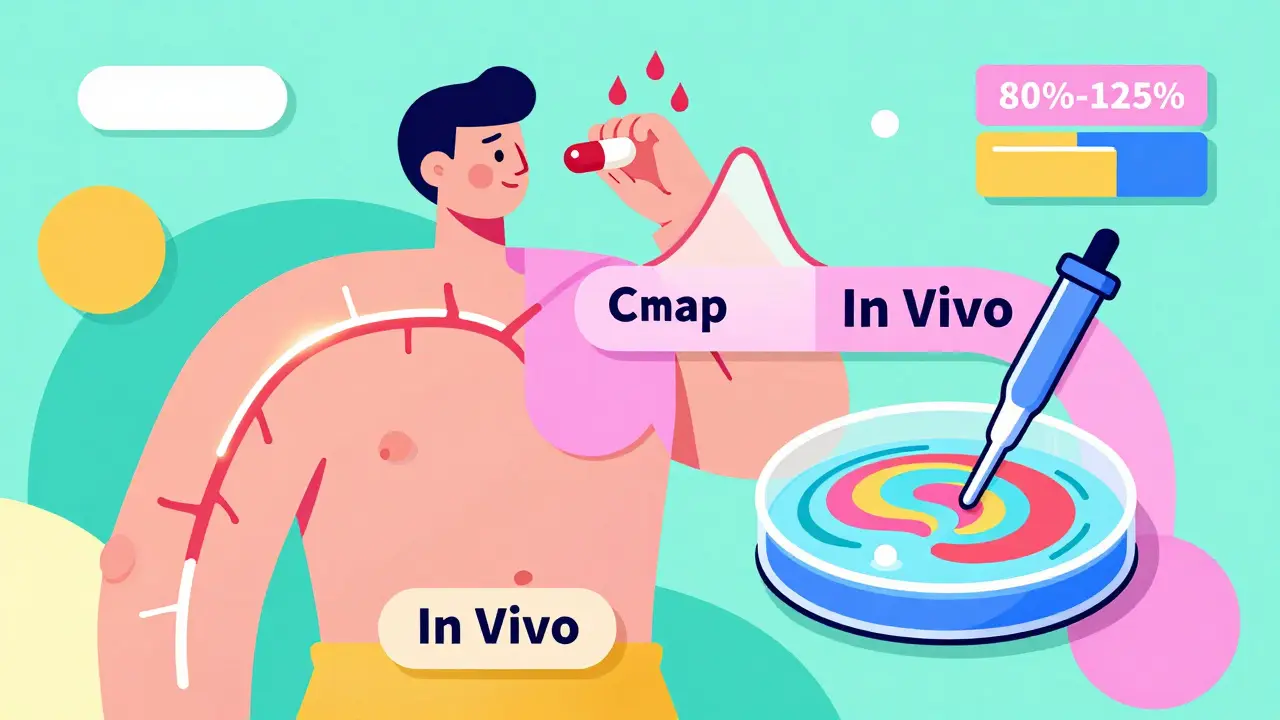
Bioequivalence testing isn’t about whether a drug looks or tastes the same. It’s about whether it delivers the same amount of active ingredient into your bloodstream at the same rate as the original. The FDA requires this proof before allowing a generic drug to be sold. Without it, a generic could be too weak to work-or too strong and cause dangerous side effects.
The standard measure? Two key numbers: Cmax (the highest concentration of drug in your blood) and AUC (the total amount of drug absorbed over time). For two products to be considered bioequivalent, the 90% confidence interval of their ratio must fall between 80% and 125%. That means the generic can’t be more than 20% weaker or stronger than the brand-name drug in how fast and how much it gets absorbed.
This isn’t just paperwork. It’s safety. For drugs like warfarin or levothyroxine, even a 5% difference can lead to blood clots or thyroid crashes. That’s why the rules aren’t one-size-fits-all.
In Vivo Testing: The Human Trial Approach
In vivo bioequivalence testing means testing in living humans. The gold standard is a two-period, two-sequence crossover study with 24 healthy volunteers. Each person takes the generic drug in one period and the brand-name drug in another, with a washout period in between. Blood samples are drawn over 24 to 72 hours to measure drug levels.
This method is direct. It shows exactly how the body handles the drug. But it’s expensive, slow, and ethically complex. A typical in vivo study costs between $500,000 and $1 million and takes 3 to 6 months to complete. It requires certified clinical units, strict FDA compliance (21 CFR Part 11), and ethical approvals.
Why do companies still use it? Because it’s the most reliable way to confirm performance for certain drugs. The FDA requires in vivo testing when:
- The drug has a narrow therapeutic index (like digoxin or phenytoin)
- The drug’s absorption is affected by food (like itraconazole or atazanavir)
- The drug has nonlinear pharmacokinetics (where dose changes don’t predict blood levels)
- The drug acts locally in the gut (like budesonide for Crohn’s disease)
Even for simple pills, if the drug doesn’t fit neatly into the Biopharmaceutics Classification System (BCS), in vivo testing is the fallback. In 2020, 95% of generic oral solid dosage forms used this method. It’s the safety net.
In Vitro Testing: The Lab-Based Alternative
In vitro bioequivalence testing skips humans entirely. Instead, scientists use lab tools to measure physical and chemical properties of the drug product. Think of it like testing a car’s engine without driving it-by checking fuel flow, valve timing, and compression ratios.
The most common method is dissolution testing. The drug is placed in a machine that mimics stomach and intestinal fluids at different pH levels (from acidic stomach to neutral intestine). The goal? To show the generic dissolves at the same rate and extent as the brand-name drug. Other methods include particle size analysis, droplet size measurement for inhalers, and content uniformity checks.
These tests are precise. Dissolution testing has a coefficient of variation (CV) under 5%, compared to 10-20% in human studies. That means less noise, more clarity.
The FDA accepts in vitro testing as sufficient for bioequivalence in specific cases:
- BCS Class I drugs (high solubility, high permeability)-like atenolol or ranitidine
- Topical products where absorption into the bloodstream isn’t the goal (like hydrocortisone cream)
- Inhalers and nasal sprays where delivery to the lungs or nasal passages matters more than blood levels
- When a validated in vitro-in vivo correlation (IVIVC) exists
For BCS Class I drugs, the FDA granted 78% of biowaiver requests in 2021 using in vitro data alone. That’s a huge win for manufacturers. One Teva scientist reported saving $1.2 million and 8 months by switching from an in vivo study to a validated dissolution method.

When In Vitro Fails and In Vivo Is Still Needed
In vitro testing isn’t magic. It can’t replicate the messy reality of the human body. Gastric emptying, enzyme activity, gut microbiome, and food interactions are impossible to fully simulate in a lab.
That’s why in vitro methods only work for certain drugs. For BCS Class III drugs (high solubility, low permeability), in vitro testing correctly predicted bioequivalence in only 65% of cases. That’s a 35% failure rate. For drugs like metformin or acyclovir, where absorption depends on transporters in the gut wall, human testing remains essential.
Even when a product gets approved based on in vitro data, problems can emerge later. A Mylan (now Viatris) regulatory manager shared that a topical antifungal approved via in vitro testing later needed an in vivo study after patients reported inconsistent results. The post-marketing study cost $850,000 and delayed expansion by 11 months.
For complex products-like nasal sprays or inhalers-regulators often require both. In 2022, 63% of nasal spray applications needed both in vitro and in vivo data. Why? Because in vitro can prove the device delivers the right dose, but only in vivo can confirm that dose actually gets absorbed in the right way.
The Future: Hybrid Approaches and New Tech
The field is shifting. The FDA’s 2023 draft guidance on nasal sprays and inhalers says in vitro testing alone can be enough-if it’s backed by physiologically relevant methods. Teva’s generic budesonide nasal spray, approved in October 2022, was the first major drug approved this way.
Now, modeling is stepping in. Physiologically based pharmacokinetic (PBPK) models simulate how a drug moves through the body using real human physiology data. The FDA accepted PBPK modeling for a modified-release product in 2023. This isn’t science fiction-it’s becoming standard.
By 2025, the FDA plans to issue two new guidances on in vitro testing for complex products. The goal? To make in vitro the default for most generics, with in vivo reserved for high-risk cases.
But don’t expect full replacement. Lawrence Lesko, former FDA clinical pharmacology director, warned: “In vitro methods can’t replicate the interplay of gut motility, pH shifts, and enzyme activity.” For drugs with absorption windows-like some antibiotics-human testing still wins.
Cost, Time, and Real-World Trade-offs
Let’s cut through the science and look at the bottom line.
In vitro testing costs $50,000-$150,000 and takes 2-4 weeks. In vivo? $500,000-$1 million and 3-6 months. That’s a 10x cost difference and a 10x time difference.
But here’s the catch: in vitro method development takes time. Setting up a dissolution method that satisfies the FDA can take 3-12 months. It requires deep expertise in analytical chemistry, biopharmaceutics, and regulatory science. Most labs need 6-12 months of training to get it right.
Meanwhile, in vivo studies need clinical sites, ethics boards, patient recruitment, and data systems compliant with 21 CFR Part 11. That’s a whole different operation.
For manufacturers, the choice isn’t just science-it’s strategy. If you’re making a BCS Class I tablet, go in vitro. Save money. Speed to market. But if your drug is tricky-low permeability, food effects, narrow window-don’t risk it. In vivo is the only way to be sure.
Global Trends and Regulatory Alignment
The U.S. isn’t alone. The European Medicines Agency approved 214 biowaivers based on in vitro data in 2022-a 27% jump from 2020. Japan and the EU now accept the same standards as the FDA for BCS Class I drugs. Harmonization is real.
The global bioequivalence testing market hit $2.1 billion in 2022. In vitro methods made up 38% of that-and are projected to hit 45% by 2028. That growth isn’t random. It’s driven by better science, better tools, and smarter regulation.
The FDA’s 2020-2025 Strategic Plan explicitly says: “Advance novel approaches to demonstrate bioequivalence.” That includes in vitro methods and model-informed decisions. The future isn’t just about doing less testing. It’s about doing smarter testing.
Can a generic drug be approved without any human testing?
Yes, but only under specific conditions. For BCS Class I drugs-those with high solubility and high permeability-regulators like the FDA and EMA accept bioequivalence based solely on in vitro testing, such as dissolution profiles. This is called a biowaiver. Over 78% of BCS Class I drug applications in 2021 were approved this way. In vitro methods are also accepted for some inhalers, nasal sprays, and topical products where systemic absorption isn’t the goal. However, for drugs with narrow therapeutic indices, food effects, or complex delivery systems, human testing (in vivo) is still required.
Why is in vivo testing still used if in vitro is cheaper and faster?
Because not all drugs behave the same way in the lab as they do in the body. In vitro tests measure physical properties like dissolution, but they can’t replicate how food, gut enzymes, pH changes, or gut motility affect drug absorption. For drugs with low permeability (BCS Class III), food interactions, or nonlinear pharmacokinetics, in vivo testing remains the only reliable way to prove the generic will work the same way in patients. The FDA still requires in vivo studies for 95% of oral solid dosage forms, especially when the drug’s absorption is unpredictable.
What’s the difference between BCS Class I and Class III drugs?
The Biopharmaceutics Classification System (BCS) groups drugs based on solubility and permeability. Class I drugs are highly soluble and highly permeable-like atenolol or ranitidine. These are ideal for in vitro testing because they’re reliably absorbed in the gut. Class III drugs are highly soluble but poorly permeable-like metformin or acyclovir. Their absorption depends on transporters in the gut wall, which in vitro tests can’t mimic well. That’s why in vitro methods work for 92% of Class I drugs but only 65% of Class III drugs.
How does the FDA decide whether to accept in vitro data?
The FDA evaluates in vitro data based on three things: the drug’s properties (like BCS class), the validity of the testing method, and whether there’s a proven correlation between lab results and human performance (IVIVC). For example, a dissolution method that matches the brand-name drug across multiple pH levels (1.2, 4.5, 6.8) and shows 90% release within 30 minutes may be accepted for immediate-release tablets. For inhalers, cascade impactor data proving consistent particle size and delivery is key. If the method is scientifically sound and validated, the FDA will accept it-even if it’s not the traditional human study.
Are in vitro methods becoming the new standard for all generics?
Not yet, but the trend is clear. The FDA, EMA, and other agencies are pushing toward in vitro as the default for simple, well-understood drugs-especially BCS Class I and certain complex delivery systems. The goal is to replace human studies where science allows it. By 2025, the FDA plans to issue new guidances to expand in vitro use. But for high-risk drugs-those with narrow therapeutic windows, food effects, or unknown absorption patterns-in vivo testing will remain mandatory. The future is hybrid: in vitro for most, in vivo only when necessary.

15 Comments
Darren Torpey
March 1, 2026 AT 11:26Man, I love how we’re finally moving away from throwing humans into clinical trials just to test pills. In vitro isn’t just cheaper-it’s smarter. We’ve got the tech now to simulate gut pH, dissolution rates, even transporter activity. Why risk a volunteer’s sleep schedule when a robot in a lab can do it better? 🚀
Lebogang kekana
March 2, 2026 AT 06:36Let me tell you something-this isn’t science, it’s capitalism in a lab coat. They want to cut costs so bad they’ll skip human testing for drugs that could kill someone if they’re off by 3%. I’ve seen people on generic warfarin turn purple. This ain’t progress, it’s negligence wrapped in a FDA memo.
Jessica Chaloux
March 3, 2026 AT 17:01OMG I just read this and I’m crying 😭 I had no idea how much goes into generics. Like… I thought they just copied the pill. But the science? The precision? The fact that a drug can be 20% weaker and still be legal?? I’m shook. Thank you for explaining this so clearly.
marjorie arsenault
March 5, 2026 AT 07:25It’s so important to remember that behind every generic drug approval is a team of scientists working late nights, running dissolution tests, and double-checking every curve. This isn’t about cutting corners-it’s about using the right tool for the job. And sometimes, that tool is a machine, not a human volunteer.
For BCS Class I drugs? In vitro is perfect. For metformin? Stick with the human trial. We’re not replacing in vivo-we’re refining when to use it.
Deborah Dennis
March 6, 2026 AT 21:45So… let me get this straight. You’re telling me we’re allowing drugs to go to market without ever testing them on actual humans? And we call this ‘science’? You know what’s in my gut? Bacteria. Stress. Coffee. Ate a burrito last night. No lab can replicate that. This is dangerous. And lazy. And frankly, terrifying.
Shivam Pawa
March 7, 2026 AT 12:06BCS Class I = high solubility high permeability = in vitro is reliable. Class III = high solubility low permeability = transporter dependent = in vivo still needed. The data is clear. The FDA guidelines are sound. The resistance is emotional, not scientific.
Also, dissolution testing CV <5% vs human CV 10-20%. That’s not noise-that’s control.
Diane Croft
March 9, 2026 AT 02:51As someone who’s been on generic levothyroxine for 12 years, I can’t tell you how much this matters. I’ve had brands switch and my TSH go haywire. That’s why I’m glad they still require in vivo for narrow-window drugs. We’re not against innovation-we’re against risk.
Donna Zurick
March 10, 2026 AT 10:19This is why I love pharma. It’s not just about making pills. It’s about matching science to real life. Sometimes the lab wins. Sometimes the body wins. And sometimes, you need both to make sure nobody gets hurt.
Tobias Mösl
March 10, 2026 AT 14:15Let’s be real. The FDA’s ‘in vitro first’ push is just Big Pharma’s way of outsourcing safety. Who’s testing these dissolution methods? The same labs that got caught falsifying data in 2018? The same companies that bribed inspectors? We’re trading human trials for corporate audits. And we’re calling it innovation. Wake up.
They approved a generic inhaler using in vitro data. Then patients started dying from under-dosing. Why? Because the lab didn’t account for how the patient’s asthma attack changed their inhalation pattern. No machine can simulate panic.
tatiana verdesoto
March 10, 2026 AT 17:08Thank you for this. I work in a clinic and I see patients every week who can’t afford brand-name meds. Knowing that there’s a solid science behind generics-not just ‘same pill, cheaper’-makes me feel better about prescribing them. It’s not about cutting corners. It’s about cutting waste.
Ethan Zeeb
March 12, 2026 AT 02:06People act like in vivo is the only real science. But you don’t need to inject a human to prove dissolution. You need to prove consistency. And dissolution testing is more consistent than a 24-person crossover study with sleep-deprived volunteers who drank coffee before the test.
The real problem? We treat every drug like it’s warfarin. It’s not. We need tiered approaches. And we’re getting there.
Aisling Maguire
March 12, 2026 AT 22:10So… if a drug’s BCS Class I, you can skip the human study? That’s wild. I thought we needed bodies. Turns out we just needed good chemistry. And a really fancy beaker.
Full Scale Webmaster
March 14, 2026 AT 03:08Let me break this down for you like you’re five: In vitro = lab. In vivo = real life. You think a machine can mimic a human stomach? Nah. You think a 30-minute dissolution test can predict how a 72-year-old with diabetes and Crohn’s absorbs a drug? Please. The FDA is selling us snake oil under the name ‘science.’
They’re doing this because they’re scared of lawsuits. If a drug kills someone because they skipped the human trial, it’s ‘regulatory innovation.’ But if they did the trial and someone died? That’s a $500M lawsuit.
And don’t get me started on how Teva’s ‘$1.2M savings’ is just a cost-cutting headline. The real cost? The patient who got the wrong dose. You don’t see that on the balance sheet.
They’re not saving time. They’re saving money. And we’re all the guinea pigs.
Angel Wolfe
March 15, 2026 AT 10:53Who owns the dissolution labs? Who certifies the methods? Who audits the data? It’s all Big Pharma insiders. The FDA’s ‘validation’ is a rubber stamp. I’ve seen the reports. The same lab does the test for the brand and the generic. Same equipment. Same technician. Same coffee stain on the notebook.
This isn’t bioequivalence. It’s collusion.
Sophia Rafiq
March 16, 2026 AT 21:13PBPK modeling is the future. Simulating absorption using real human physiology data? That’s next-level. And yes, the FDA approved it in 2023. It’s not sci-fi-it’s peer-reviewed. The real innovation isn’t skipping humans. It’s understanding them better than ever before.